- THERMODYNAMIQUE - Thermodynamique chimique
- THERMODYNAMIQUE - Thermodynamique chimiqueOn peut dire qu’au milieu du siècle dernier les bases fondamentales de la thermodynamique classique et de la théorie de l’énergie étaient établies grâce aux travaux de V. Hess, S. Carnot, J. R. von Mayer, J. P. Joule, R. J. E. Clausius, lord Kelvin. Les lois ainsi mises au jour contribuèrent puissamment au développement des machines thermiques, mais leur rigueur et leur généralité incitèrent à les appliquer à toutes les modifications des corps qui nous entourent.Les applications à la chimie prirent naissance avec les travaux de Clausius et d’Émile Clapeyron, mais c’est seulement dans le dernier quart du siècle dernier qu’elles se développèrent. La personnalité de J. W. Gibbs s’y détache et, grâce à son introduction systématique du potentiel chimique, l’évolution se poursuit de façon claire et logique.Ces recherches, essentiellement théoriques, furent utilisées ultérieurement par des chercheurs de renom: H. W. B. Roozeboom (règle des phases), Van Laar, P. Duhem (applications du potentiel chimique). Enfin, G. N. Lewis simplifia le développement des calculs, en introduisant (1901) les notions de fugacité et d’activité, qui permettent, pour les gaz non parfaits et les solutions quelconques, d’exprimer le potentiel chimique des constituants de façon aussi simple que pour un gaz parfait.Cependant, ces travaux se préoccupent peu de la réaction chimique elle-même; les grandeurs envisagées se rapportent soit au système total, soit à l’un des constituants, mais non à la réaction (chaleur de réaction, travail maximal). Pourtant, l’intérêt de la thermodynamique pour le chimiste avait été pressenti dès 1866 par H. Sainte-Claire Deville, et aussi par Horstmann (1869), mais ce sont surtout H. L. Helmholtz, J. H. Van’t Hoff et W. Nerst qui s’y attachèrent, et, au début du siècle, les chimistes généralisent son emploi. On retrouve à cette époque (1905), dans un remarquable travail de F. Haber, les données nécessaires au calcul des enthalpies libres de réactions.Malgré les progrès indéniables qu’elle a introduits dans l’étude de la réaction chimique, la précédente conception n’envisage guère que les états d’équilibre et les transformations réversibles, alors qu’une réaction chimique est manifestement irréversible et qu’il est fondamental de prévoir si elle est possible. C’est pourquoi T. De Donder, depuis 1920, a introduit l’«affinité chimique», grandeur caractéristique de l’irréversibilité.Ainsi, une thermodynamique chimique des états d’équilibre s’est substituée progressivement aux lois déjà connues de la thermochimie, mais avec une interprétation directement déduite des deux grands principes de la thermodynamique. D’où la dénomination actuelle.1. Équilibres chimiques et prévision des réactionsSoit une réaction du type:
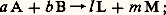 après avoir attendu le temps suffisant pour qu’elle se produise (elle n’est pas obligatoirement instantanée), on dit qu’elle est complète si le réactif de départ en proportion la plus faible (les proportions employées n’étant pas obligatoirement celles de la réaction) a été consommé entièrement.Dans le cas contraire, on dit qu’il y a équilibre: les produits d’arrivée et de départ coexistent alors, leurs proportions n’évoluant plus au cours du temps. Si A et B ne peuvent réagir complètement, c’est qu’au-delà d’un certain avancement de la réaction la recombinaison de L et de M s’y oppose. Un équilibre implique donc que, si A et B sont susceptibles de réagir de façon limitée, il en soit de même de L et de M, ce qu’on écrit:
après avoir attendu le temps suffisant pour qu’elle se produise (elle n’est pas obligatoirement instantanée), on dit qu’elle est complète si le réactif de départ en proportion la plus faible (les proportions employées n’étant pas obligatoirement celles de la réaction) a été consommé entièrement.Dans le cas contraire, on dit qu’il y a équilibre: les produits d’arrivée et de départ coexistent alors, leurs proportions n’évoluant plus au cours du temps. Si A et B ne peuvent réagir complètement, c’est qu’au-delà d’un certain avancement de la réaction la recombinaison de L et de M s’y oppose. Un équilibre implique donc que, si A et B sont susceptibles de réagir de façon limitée, il en soit de même de L et de M, ce qu’on écrit: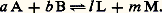 Il semble que ce soit Claude Berthollet qui ait considéré, dès 1803, que les réactions inverses que représente une équation chimique sont limitées l’une par l’autre, de sorte que le système de départ évolue vers ce qu’il appelle déjà un «état d’équilibre» où tous les participants sont présents en quantités déterminées.Cependant, ce n’est que soixante ans plus tard que les expériences de Marcelin Berthelot et Péan de Saint-Gilles (estérification, 1862) et de Sainte-Claire Deville (dissociation thermique de la vapeur d’eau, 1863) permirent d’étudier systématiquement l’équilibre chimique.La première relation quantitative entre les proportions des corps en présence à l’équilibre est la loi d’action de masse, due à C. Guldberg et P. Waage (1867), qui n’était fondée que sur des considérations empiriques, mais qui s’est révélée exacte par la suite (Hortsmann, 1873). Si l’on y adjoint la relation de Van’t Hoff (1884) donnant la variation de la constante d’action de masse avec la température, on dispose des moyens nécessaires pour prévoir les variations des proportions à l’équilibre en fonction des conditions (température, pression, proportions des réactifs). Ces lois du «déplacement de l’équilibre » par variations de ces facteurs ont été énoncées de façon qualitative mais extrêmement générale par H. Le Chatelier (1885) et K. F. Braun (1886). Enfin, grâce aux travaux de Nernst (1906) et de Max Planck (1912), les constantes d’équilibre ont pu être évaluées à partir de données uniquement calorimétriques.Si un même état d’équilibre est réalisé, que l’on parte de A + B ou de L + M (les conditions étant évidemment les mêmes), on dit qu’il y a réversibilité. En revanche, il peut arriver que les vitesses des réactions directe (A sur B) et inverse s’annulent avant d’avoir atteint les proportions requises pour le véritable équilibre: on obtient alors deux limites différentes selon les réactifs de départ; dans ce cas, il n’y a pas réversibilité.Des substances A et B susceptibles de réagir entre elles pour donner les composés L et M, selon les relations données ci-dessus, réagiront-elles réellement dans les conditions de température, de pression et de proportion des réactifs mis en jeu que nous avons adoptées? Autrement dit, auront-elles de l’affinité l’une pour l’autre?Il faut arriver à J. Thomsen (1858), puis à Berthelot (1865) pour trouver l’idée de mesure de l’affinité, qui serait donnée par la chaleur de réaction. En réalité, Helmholtz (1882) montra que c’est la variation d’enthalpie libre accompagnant la réaction éventuelle qui décide de sa possibilité.Cette définition, couramment admise, nous la développerons, mais elle n’est pas entièrement satisfaisante, car elle ne fait intervenir que les états initial et final, et non pas la façon dont on passe de l’un à l’autre. La théorie de l’affinité de T. De Donder évoquée plus haut, devait ensuite combler cette lacune.2. Potentiel chimiqueAprès Gibbs, l’emploi du potentiel chimique molaire:
Il semble que ce soit Claude Berthollet qui ait considéré, dès 1803, que les réactions inverses que représente une équation chimique sont limitées l’une par l’autre, de sorte que le système de départ évolue vers ce qu’il appelle déjà un «état d’équilibre» où tous les participants sont présents en quantités déterminées.Cependant, ce n’est que soixante ans plus tard que les expériences de Marcelin Berthelot et Péan de Saint-Gilles (estérification, 1862) et de Sainte-Claire Deville (dissociation thermique de la vapeur d’eau, 1863) permirent d’étudier systématiquement l’équilibre chimique.La première relation quantitative entre les proportions des corps en présence à l’équilibre est la loi d’action de masse, due à C. Guldberg et P. Waage (1867), qui n’était fondée que sur des considérations empiriques, mais qui s’est révélée exacte par la suite (Hortsmann, 1873). Si l’on y adjoint la relation de Van’t Hoff (1884) donnant la variation de la constante d’action de masse avec la température, on dispose des moyens nécessaires pour prévoir les variations des proportions à l’équilibre en fonction des conditions (température, pression, proportions des réactifs). Ces lois du «déplacement de l’équilibre » par variations de ces facteurs ont été énoncées de façon qualitative mais extrêmement générale par H. Le Chatelier (1885) et K. F. Braun (1886). Enfin, grâce aux travaux de Nernst (1906) et de Max Planck (1912), les constantes d’équilibre ont pu être évaluées à partir de données uniquement calorimétriques.Si un même état d’équilibre est réalisé, que l’on parte de A + B ou de L + M (les conditions étant évidemment les mêmes), on dit qu’il y a réversibilité. En revanche, il peut arriver que les vitesses des réactions directe (A sur B) et inverse s’annulent avant d’avoir atteint les proportions requises pour le véritable équilibre: on obtient alors deux limites différentes selon les réactifs de départ; dans ce cas, il n’y a pas réversibilité.Des substances A et B susceptibles de réagir entre elles pour donner les composés L et M, selon les relations données ci-dessus, réagiront-elles réellement dans les conditions de température, de pression et de proportion des réactifs mis en jeu que nous avons adoptées? Autrement dit, auront-elles de l’affinité l’une pour l’autre?Il faut arriver à J. Thomsen (1858), puis à Berthelot (1865) pour trouver l’idée de mesure de l’affinité, qui serait donnée par la chaleur de réaction. En réalité, Helmholtz (1882) montra que c’est la variation d’enthalpie libre accompagnant la réaction éventuelle qui décide de sa possibilité.Cette définition, couramment admise, nous la développerons, mais elle n’est pas entièrement satisfaisante, car elle ne fait intervenir que les états initial et final, et non pas la façon dont on passe de l’un à l’autre. La théorie de l’affinité de T. De Donder évoquée plus haut, devait ensuite combler cette lacune.2. Potentiel chimiqueAprès Gibbs, l’emploi du potentiel chimique molaire: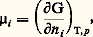 s’est progressivement imposé de préférence au potentiel chimique massique défini par l’égalité (18) de l’article THERMODYNAMIQUE - B. Lois fondamentales, et que nous noterons ici (Th. 18). Toutefois l’interprétation est similaire. C’est ainsi, par exemple, que la loi (Th. 19) exprimant l’homogénéité linéaire de l’enthalpie libre prend ici la forme symétrique:
s’est progressivement imposé de préférence au potentiel chimique massique défini par l’égalité (18) de l’article THERMODYNAMIQUE - B. Lois fondamentales, et que nous noterons ici (Th. 18). Toutefois l’interprétation est similaire. C’est ainsi, par exemple, que la loi (Th. 19) exprimant l’homogénéité linéaire de l’enthalpie libre prend ici la forme symétrique: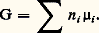 Pour une transformation où l’on fait varier arbitrairement le nombre de moles à température et à pression constantes, on a:
Pour une transformation où l’on fait varier arbitrairement le nombre de moles à température et à pression constantes, on a: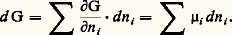 Puisque l’on a posé au chapitre 3 de l’article THERMODYNAMIQUE - B. Lois fondamentales:
Puisque l’on a posé au chapitre 3 de l’article THERMODYNAMIQUE - B. Lois fondamentales: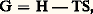 on peut écrire:
on peut écrire: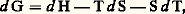 soit (Th. 17):
soit (Th. 17):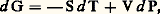 d’où:
d’où: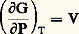 (formule valable pour tout système en équilibre).Compte tenu de la relation PV = n RT, si le gaz est parfait, on peut tirer:
(formule valable pour tout système en équilibre).Compte tenu de la relation PV = n RT, si le gaz est parfait, on peut tirer: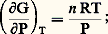 en intégrant G = G0 + n RT ln P, soit par mole 猪 = 猪0 + RT ln P, où le potentiel chimique 猪0, c’est-à-dire G0/n , peut dépendre de T, car l’intégration est faite à température constante.Si le gaz i appartient à un mélange de gaz parfaits dans lequel les interactions sont négligeables, on peut considérer que ses propriétés sont indépendantes des autres constituants, et l’on appelle pression partielle p i celle qu’il aurait s’il occupait à lui seul tout le volume p i = n i RT/V à la même température.Par analogie, on aura ainsi:
en intégrant G = G0 + n RT ln P, soit par mole 猪 = 猪0 + RT ln P, où le potentiel chimique 猪0, c’est-à-dire G0/n , peut dépendre de T, car l’intégration est faite à température constante.Si le gaz i appartient à un mélange de gaz parfaits dans lequel les interactions sont négligeables, on peut considérer que ses propriétés sont indépendantes des autres constituants, et l’on appelle pression partielle p i celle qu’il aurait s’il occupait à lui seul tout le volume p i = n i RT/V à la même température.Par analogie, on aura ainsi: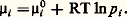 On peut remarquer que, si l’on désigne par concentration c i le nombre de moles par unité de volume, on obtient p i = c i RT, soit, en introduisant dans 猪0 tout ce qui dépend de T,
On peut remarquer que, si l’on désigne par concentration c i le nombre de moles par unité de volume, on obtient p i = c i RT, soit, en introduisant dans 猪0 tout ce qui dépend de T,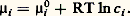 Si l’espèce i et celles qui l’accompagnent (solutés) sont dissoutes en concentrations suffisamment faibles dans un solvant, il n’y a d’interactions qu’avec les molécules de ce dernier. On considère que l’espèce i se comporte dans le solvant comme le ferait un gaz parfait (dans le vide) à la même concentration et on applique l’équation précédente.Cependant, lorsque c i ou les concentrations des autres solutés croissent, le comportement se complique et 猪i devient de la forme:
Si l’espèce i et celles qui l’accompagnent (solutés) sont dissoutes en concentrations suffisamment faibles dans un solvant, il n’y a d’interactions qu’avec les molécules de ce dernier. On considère que l’espèce i se comporte dans le solvant comme le ferait un gaz parfait (dans le vide) à la même concentration et on applique l’équation précédente.Cependant, lorsque c i ou les concentrations des autres solutés croissent, le comportement se complique et 猪i devient de la forme: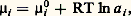 où a i est l’activité: c’est la concentration fictive que devrait adopter l’espèce i pour continuer à présenter le comportement des solutions diluées. En solution suffisamment diluée, on a a i 力 c i , mais, aux concentrations plus élevées, l’écart est notable.L’état standard d’un composé est l’état de ce composé pur à la température considérée, sous la pression de 1 atmosphère. S’il est en solution, son activité doit être égale à 1 (ou sa concentration si, pour 1 mole/litre, il obéit encore aux lois des solutions diluées).Dans les formules précédentes, les 猪0 représentent donc des potentiels chimiques standards.Équilibre entre plusieurs phasesOn appelle variance d’un système hétérogène (à plusieurs phases) le nombre minimal de variables intensives nécessaires pour fixer son état, c’est-à-dire les valeurs des autres variables.Les constituants d’un système chimique ne sont pas indépendants, car l’un d’entre eux peut prendre naissance à partir des autres, par suite de la réaction chimique; on appelle constituants indépendants le nombre de constituants, diminué du nombre de réactions en équilibre possibles. Ainsi dans la réaction CaC3 燎 CaO + C2 il n’y a que deux constituants indépendants.La règle des phases, ou règle de Gibbs, sert à fixer la variance. Soit n le nombre des constituants indépendants et 﨏 le nombre de phases. La composition de chaque phase serait définie si l’on connaissait les proportions des constituants, soit (n 漣 1) quantités inconnues (la dernière se déduit des autres, puisque la somme des proportions est égale à 1); au total, il faut connaître 﨏(n 漣 1) quantités, auxquelles il faut ajouter les variables intensives habituelles T et P.Cependant, il a été démontré que l’équilibre entre deux phases suppose que le potentiel chimique 猪i possède la même valeur pour chaque constituant dans les deux phases. Cette condition est généralisable à plus de deux phases; cela donne ( 﨏 漣 1) égalités, donc ( 﨏 漣 1) relations entre les grandeurs dont dépend 猪i . Au total, pour les n constituants, on obtient n ( 﨏 漣 1) relations entre ces grandeurs.Il en résulte que la variance a pour valeur:
où a i est l’activité: c’est la concentration fictive que devrait adopter l’espèce i pour continuer à présenter le comportement des solutions diluées. En solution suffisamment diluée, on a a i 力 c i , mais, aux concentrations plus élevées, l’écart est notable.L’état standard d’un composé est l’état de ce composé pur à la température considérée, sous la pression de 1 atmosphère. S’il est en solution, son activité doit être égale à 1 (ou sa concentration si, pour 1 mole/litre, il obéit encore aux lois des solutions diluées).Dans les formules précédentes, les 猪0 représentent donc des potentiels chimiques standards.Équilibre entre plusieurs phasesOn appelle variance d’un système hétérogène (à plusieurs phases) le nombre minimal de variables intensives nécessaires pour fixer son état, c’est-à-dire les valeurs des autres variables.Les constituants d’un système chimique ne sont pas indépendants, car l’un d’entre eux peut prendre naissance à partir des autres, par suite de la réaction chimique; on appelle constituants indépendants le nombre de constituants, diminué du nombre de réactions en équilibre possibles. Ainsi dans la réaction CaC3 燎 CaO + C2 il n’y a que deux constituants indépendants.La règle des phases, ou règle de Gibbs, sert à fixer la variance. Soit n le nombre des constituants indépendants et 﨏 le nombre de phases. La composition de chaque phase serait définie si l’on connaissait les proportions des constituants, soit (n 漣 1) quantités inconnues (la dernière se déduit des autres, puisque la somme des proportions est égale à 1); au total, il faut connaître 﨏(n 漣 1) quantités, auxquelles il faut ajouter les variables intensives habituelles T et P.Cependant, il a été démontré que l’équilibre entre deux phases suppose que le potentiel chimique 猪i possède la même valeur pour chaque constituant dans les deux phases. Cette condition est généralisable à plus de deux phases; cela donne ( 﨏 漣 1) égalités, donc ( 﨏 漣 1) relations entre les grandeurs dont dépend 猪i . Au total, pour les n constituants, on obtient n ( 﨏 漣 1) relations entre ces grandeurs.Il en résulte que la variance a pour valeur: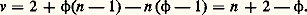 3. Grandeurs caractéristiques d’une réactionChaleur de réactionSoit une réaction du type: l’effet thermique accompagnant la réaction, à pression constante, de a moles de la substance A sur b moles de B pour donner l moles de L + m moles M s’appelle chaleur de réaction à pression constante notée Q.On voit (Th. 3), en faisant d P = 0, qu’une intégration immédiate entre états final et initial donne:
3. Grandeurs caractéristiques d’une réactionChaleur de réactionSoit une réaction du type: l’effet thermique accompagnant la réaction, à pression constante, de a moles de la substance A sur b moles de B pour donner l moles de L + m moles M s’appelle chaleur de réaction à pression constante notée Q.On voit (Th. 3), en faisant d P = 0, qu’une intégration immédiate entre états final et initial donne: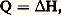 ce qui donne un sens physique à l’enthalpie H.Comme H est une fonction d’état, H (donc Q) ne dépend que des états initial et final parmi l’ensemble des transformations isobares. De même (Th. 2), pour les isomètres (d V = 0), Q devient une fonction d’état: Q = E (lois de Hess).La variation d’enthalpie H est évaluée (en particulier par calorimétrie) à la température ordinaire. Si la réaction est étudiée à température élevée, il est nécessaire de connaître H en fonction de T.Pour cela, appliquons la relation (Th. 3):
ce qui donne un sens physique à l’enthalpie H.Comme H est une fonction d’état, H (donc Q) ne dépend que des états initial et final parmi l’ensemble des transformations isobares. De même (Th. 2), pour les isomètres (d V = 0), Q devient une fonction d’état: Q = E (lois de Hess).La variation d’enthalpie H est évaluée (en particulier par calorimétrie) à la température ordinaire. Si la réaction est étudiée à température élevée, il est nécessaire de connaître H en fonction de T.Pour cela, appliquons la relation (Th. 3):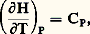 aux états initial et final. Il vient l’équation de Kirchhoff:
aux états initial et final. Il vient l’équation de Kirchhoff: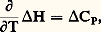 où CP désigne la différence entre les capacités thermiques des produits d’arrivée et de départ.Si Ci désigne la capacité thermique molaire de chaque constituant, on a:
où CP désigne la différence entre les capacités thermiques des produits d’arrivée et de départ.Si Ci désigne la capacité thermique molaire de chaque constituant, on a: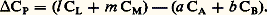 L’intégration de cette équation entre la température ordinaire (généralement 25 0C, soit 298 K) et la température envisagée permet de connaître H pour cette dernière si les Ci sont connus en fonction de T:
L’intégration de cette équation entre la température ordinaire (généralement 25 0C, soit 298 K) et la température envisagée permet de connaître H pour cette dernière si les Ci sont connus en fonction de T: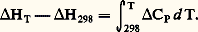 Enthalpie libre de réactionL’enthalpie libre de réaction est la variation d’enthalpie libre G accompagnant une réaction a A + b Bl L + m M, effectuée dans les conditions où se trouve le système, en ce qui concerne la température, la pression totale et les proportions des réactifs.Il faut donc calculer la variation GT,P lorsque, à une température T et à une pression P données, A et B réagissent selon leurs coefficients de réaction pour donner L et M; on supposera le système en forte quantité, afin que la transformation ne perturbe pas sa composition.La formule d G = 猪i dn i donne, avec n A = 漣 a , n B = 漣 b , n L = l et n M = m , la relation suivante:
Enthalpie libre de réactionL’enthalpie libre de réaction est la variation d’enthalpie libre G accompagnant une réaction a A + b Bl L + m M, effectuée dans les conditions où se trouve le système, en ce qui concerne la température, la pression totale et les proportions des réactifs.Il faut donc calculer la variation GT,P lorsque, à une température T et à une pression P données, A et B réagissent selon leurs coefficients de réaction pour donner L et M; on supposera le système en forte quantité, afin que la transformation ne perturbe pas sa composition.La formule d G = 猪i dn i donne, avec n A = 漣 a , n B = 漣 b , n L = l et n M = m , la relation suivante: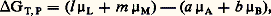 soit, pour un gaz, en remplaçant les 猪i par leurs valeurs,
soit, pour un gaz, en remplaçant les 猪i par leurs valeurs,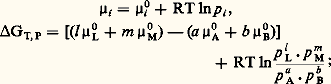 dans cette dernière formule, le terme entre crochets ne dépend que de T (comme les 猪0) et représente G0, enthalpie libre standard de réaction, c’est-à-dire G lorsque A et B réagissent dans leur état standard (p = 1 atm.) pour donner L et M dans leur état standard.Si la réaction a lieu en solution, on obtient une expression analogue en substituant les a i (ou les c i ) aux p i .Entropie de réactionOn appelle entropie de réaction la variation S d’entropie accompagnant une réaction, soit:
dans cette dernière formule, le terme entre crochets ne dépend que de T (comme les 猪0) et représente G0, enthalpie libre standard de réaction, c’est-à-dire G lorsque A et B réagissent dans leur état standard (p = 1 atm.) pour donner L et M dans leur état standard.Si la réaction a lieu en solution, on obtient une expression analogue en substituant les a i (ou les c i ) aux p i .Entropie de réactionOn appelle entropie de réaction la variation S d’entropie accompagnant une réaction, soit: Cette grandeur est liée à la chaleur de réaction et à l’enthalpie libre de réaction, car de l’expression G = H 漣 TS on déduit pour une température T donnée:
Cette grandeur est liée à la chaleur de réaction et à l’enthalpie libre de réaction, car de l’expression G = H 漣 TS on déduit pour une température T donnée: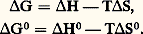 Les entropies des substances, de même que H, étant accessibles par mesures de CP (Th. 12), donc par calorimétrie, on dispose ainsi d’un moyen de détermination de G par calorimétrie uniquement.Enthalpies et enthalpies libres standards de formationOn appelle enthalpie de formation d’un composé sa chaleur de formation à pression constante à partir de ses éléments; ainsi, pour la réaction:
Les entropies des substances, de même que H, étant accessibles par mesures de CP (Th. 12), donc par calorimétrie, on dispose ainsi d’un moyen de détermination de G par calorimétrie uniquement.Enthalpies et enthalpies libres standards de formationOn appelle enthalpie de formation d’un composé sa chaleur de formation à pression constante à partir de ses éléments; ainsi, pour la réaction: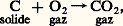 l’enthalpie de formation du dioxyde de carbone est H = 漣 382,9 kJ.On a dressé des tables d’enthalpies standards de formation (généralement à 25 0C), car leur connaissance permet de calculer la variation d’enthalpie de toute réaction.L’enthalpie H étant une fonction d’état, sa variation H ne dépend que des états initial et final. On peut donc décomposer la réaction en deux étapes: dans la première, on dissocie les produits réagissants en leurs éléments, ce qui libère leurs enthalpies de formation changées de signe; dans la seconde on recombine les éléments pour donner les produits d’arrivée, ce qui libère leurs enthalpies de formation. L’enthalpie de réaction est la somme de ces deux opérations et est égale aux différences des enthalpies de formation des produits d’arrivée et de départ. Les mêmes considérations s’appliquent aux enthalpies libres.On a dressé également des tables d’entropies standards; contrairement aux tables d’enthalpies, dont la grandeur envisagée est donnée par rapport aux enthalpies des éléments (ce qui n’entrave en rien leur utilisation), les entropies standards sont obtenues en valeur absolue. En effet, alors que les enthalpies ou les enthalpies libres standards de formation des éléments sont évidemment nulles, leurs entropies standards, mesurées à partir des capacités thermiques, ne le sont pas.4. Les équilibres chimiquesLa connaissance de la variation d’enthalpie libre G est fondamentale, car, à T et à P données, si, dans les conditions de l’expérience, cette variation est négative, les composants du système réagiront (cf. THERMODYNAMIQUE - Lois fondamentales). En revanche, si en fonction des conditions opératoires, la valeur de G est minimale, elle ne pourra diminuer: le système n’évoluera pas, et il sera donc en équilibre; la condition d’équilibre s’écrira, par suite, en exprimant GT,P pour une transformation possible et en écrivant GT,P = 0.Conditions d’équilibreLa condition précédente appliquée à la variation GT,P de réaction donne, en posant G0 = 漣 RT ln KP,
l’enthalpie de formation du dioxyde de carbone est H = 漣 382,9 kJ.On a dressé des tables d’enthalpies standards de formation (généralement à 25 0C), car leur connaissance permet de calculer la variation d’enthalpie de toute réaction.L’enthalpie H étant une fonction d’état, sa variation H ne dépend que des états initial et final. On peut donc décomposer la réaction en deux étapes: dans la première, on dissocie les produits réagissants en leurs éléments, ce qui libère leurs enthalpies de formation changées de signe; dans la seconde on recombine les éléments pour donner les produits d’arrivée, ce qui libère leurs enthalpies de formation. L’enthalpie de réaction est la somme de ces deux opérations et est égale aux différences des enthalpies de formation des produits d’arrivée et de départ. Les mêmes considérations s’appliquent aux enthalpies libres.On a dressé également des tables d’entropies standards; contrairement aux tables d’enthalpies, dont la grandeur envisagée est donnée par rapport aux enthalpies des éléments (ce qui n’entrave en rien leur utilisation), les entropies standards sont obtenues en valeur absolue. En effet, alors que les enthalpies ou les enthalpies libres standards de formation des éléments sont évidemment nulles, leurs entropies standards, mesurées à partir des capacités thermiques, ne le sont pas.4. Les équilibres chimiquesLa connaissance de la variation d’enthalpie libre G est fondamentale, car, à T et à P données, si, dans les conditions de l’expérience, cette variation est négative, les composants du système réagiront (cf. THERMODYNAMIQUE - Lois fondamentales). En revanche, si en fonction des conditions opératoires, la valeur de G est minimale, elle ne pourra diminuer: le système n’évoluera pas, et il sera donc en équilibre; la condition d’équilibre s’écrira, par suite, en exprimant GT,P pour une transformation possible et en écrivant GT,P = 0.Conditions d’équilibreLa condition précédente appliquée à la variation GT,P de réaction donne, en posant G0 = 漣 RT ln KP,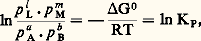 d’où la loi d’action de masse de Guldberg et Waage (1867):
d’où la loi d’action de masse de Guldberg et Waage (1867):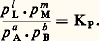 Dans le cas des gaz parfaits, la «constante» d’action de masse KP ne dépend que de la température, comme G0, et lui est liée directement, ce qui lui donne sa signification physique.En solution, une relation du même ordre est applicable aux activités ou aux concentrations a i ou c i , KP étant remplacée par Ka , constante d’action de masse appliquée aux activités.Il est souvent plus souhaitable d’obtenir une relation entre les proportions des constituants. La loi des gaz parfaits s’applique aussi bien à chaque constituant i qu’à l’ensemble:
Dans le cas des gaz parfaits, la «constante» d’action de masse KP ne dépend que de la température, comme G0, et lui est liée directement, ce qui lui donne sa signification physique.En solution, une relation du même ordre est applicable aux activités ou aux concentrations a i ou c i , KP étant remplacée par Ka , constante d’action de masse appliquée aux activités.Il est souvent plus souhaitable d’obtenir une relation entre les proportions des constituants. La loi des gaz parfaits s’applique aussi bien à chaque constituant i qu’à l’ensemble: n étant le nombre total de moles; d’où:
n étant le nombre total de moles; d’où: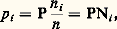 en appelant Ni la fraction molaire (ou proportion) de i , avec Ni = 1.En transposant dans la relation précédente, on obtient, en posant
en appelant Ni la fraction molaire (ou proportion) de i , avec Ni = 1.En transposant dans la relation précédente, on obtient, en posant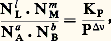 soit une relation entre les proportions des constituants, en fonction de T (par KP) et de P (par P size=1益).Cas des systèmes hétérogènesIl arrive fréquemment que, dans un système hétérogène, un constituant ou plusieurs forment une phase solide pure; un tel constituant est alors dans son état standard, et l’on a 猪i = 猪i 0; il s’ensuit qu’il n’intervient pas dans l’expression du premier membre donnant les p i ou les Ni , ainsi que dans l’évaluation de 益.Soit les trois exemples suivants de systèmes hétérogènes.
soit une relation entre les proportions des constituants, en fonction de T (par KP) et de P (par P size=1益).Cas des systèmes hétérogènesIl arrive fréquemment que, dans un système hétérogène, un constituant ou plusieurs forment une phase solide pure; un tel constituant est alors dans son état standard, et l’on a 猪i = 猪i 0; il s’ensuit qu’il n’intervient pas dans l’expression du premier membre donnant les p i ou les Ni , ainsi que dans l’évaluation de 益.Soit les trois exemples suivants de systèmes hétérogènes.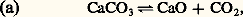 équilibre de dissociation du carbonate de calcium (solide) en chaux vive (solide) et en dioxyde de carbone (gaz); KP = p CO2 n’est donc fonction que de la température: le système est en effet monovariant, avec n = 2 et 﨏 = 3, chaque solide formant une phase distincte;
équilibre de dissociation du carbonate de calcium (solide) en chaux vive (solide) et en dioxyde de carbone (gaz); KP = p CO2 n’est donc fonction que de la température: le système est en effet monovariant, avec n = 2 et 﨏 = 3, chaque solide formant une phase distincte;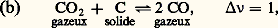 et, avec CO2 + CO = 1, on obtient 2CO/(1 漣 CO) = KP/P; le système est bivariant (n = 2 et 﨏 = 2) et l’expression précédente donne bien CO en fonction de P et de T (par l’intermédiaire de KP);
et, avec CO2 + CO = 1, on obtient 2CO/(1 漣 CO) = KP/P; le système est bivariant (n = 2 et 﨏 = 2) et l’expression précédente donne bien CO en fonction de P et de T (par l’intermédiaire de KP);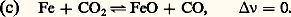 On obtient de même:
On obtient de même: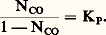 Le système est bivariant, avec n = 3 et 﨏 = 3; cependant, l’expression précédente montre que CO ne dépend que de T (par l’intermédiaire de KP), ce qui pourrait faire croire qu’il est monovariant.En réalité, pour qu’il en soit ainsi, il faudrait que toutes ses propriétés ne dépendent que de T; or, s’il en est ainsi pour CO, ce n’est pas le cas, par exemple pour p CO = CO/P qui dépend également de P.Le système 3FeO + C2 燎 Fe34 + CO se traite de même (cf. chap. 6).Variations de KP avec TLa formule de Gibbs-Helmholtz (cf. THERMODYNAMIQUE - Lois fondamentales, équation 20) s’applique aussi bien, à température constante, à l’état final qu’à l’état initial; en raisonnant sur les grandeurs standards, on obtient alors:
Le système est bivariant, avec n = 3 et 﨏 = 3; cependant, l’expression précédente montre que CO ne dépend que de T (par l’intermédiaire de KP), ce qui pourrait faire croire qu’il est monovariant.En réalité, pour qu’il en soit ainsi, il faudrait que toutes ses propriétés ne dépendent que de T; or, s’il en est ainsi pour CO, ce n’est pas le cas, par exemple pour p CO = CO/P qui dépend également de P.Le système 3FeO + C2 燎 Fe34 + CO se traite de même (cf. chap. 6).Variations de KP avec TLa formule de Gibbs-Helmholtz (cf. THERMODYNAMIQUE - Lois fondamentales, équation 20) s’applique aussi bien, à température constante, à l’état final qu’à l’état initial; en raisonnant sur les grandeurs standards, on obtient alors: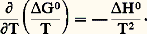 Pour une réaction chimique, on a montré que:
Pour une réaction chimique, on a montré que: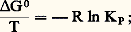 on déduit:
on déduit: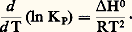 L’équation ci-dessus est également valable pour Ka .Cette relation, dite équation de Van’t Hoff , est extrêmement importante, puisqu’elle permet, à partir de la seule connaissance de la chaleur de réaction, de connaître, par intégration, KP à toute température T, à condition de la connaître à une température donnée :
L’équation ci-dessus est également valable pour Ka .Cette relation, dite équation de Van’t Hoff , est extrêmement importante, puisqu’elle permet, à partir de la seule connaissance de la chaleur de réaction, de connaître, par intégration, KP à toute température T, à condition de la connaître à une température donnée :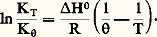 L’opération précédente suppose H0 constante; si, au contraire, elle varie avec T, on l’exprimera en fonction de T en utilisant la formule de Kirchhoff. En reportant sa valeur dans l’équation de Van’t Hoff, l’intégration de cette dernière entre T et permettra de même d’exprimer KP en fonction de T.Déplacement de l’équilibreLes proportions Ni des constituants, dans un équilibre, dépendent généralement de la température, de la pression et des proportions utilisées au départ qui ne correspondent pas obligatoirement aux coefficients de réaction. Si l’on modifie l’un de ces facteurs, l’équilibre se modifie également (on dit qu’il se déplace). Pour savoir dans quel sens il le fait, on pourra toujours calculer les nouvelles valeurs des Ni , en utilisant en particulier la formule:
L’opération précédente suppose H0 constante; si, au contraire, elle varie avec T, on l’exprimera en fonction de T en utilisant la formule de Kirchhoff. En reportant sa valeur dans l’équation de Van’t Hoff, l’intégration de cette dernière entre T et permettra de même d’exprimer KP en fonction de T.Déplacement de l’équilibreLes proportions Ni des constituants, dans un équilibre, dépendent généralement de la température, de la pression et des proportions utilisées au départ qui ne correspondent pas obligatoirement aux coefficients de réaction. Si l’on modifie l’un de ces facteurs, l’équilibre se modifie également (on dit qu’il se déplace). Pour savoir dans quel sens il le fait, on pourra toujours calculer les nouvelles valeurs des Ni , en utilisant en particulier la formule: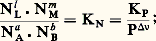 cependant, il est parfois important de savoir qualitativement, mais de façon rapide, dans quel sens l’équilibre se déplace par variation d’un facteur.Influence de la températureLes variables autres que la température étant maintenues constantes, l’équation de Van’t Hoff donne (voir ci-dessus):
cependant, il est parfois important de savoir qualitativement, mais de façon rapide, dans quel sens l’équilibre se déplace par variation d’un facteur.Influence de la températureLes variables autres que la température étant maintenues constantes, l’équation de Van’t Hoff donne (voir ci-dessus):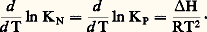 Si la variation d’enthalpie ( H) est positive (réaction endothermique), on voit que ln KN et, par suite, KN croissent par élévation de température, ce qui ne peut se faire que si L, M augmentent et si A, B diminuent: ainsi, une élévation de température déplace l’équilibre dans le sens endothermique.Influence de la pression et du volumeLes variables autres que la pression étant maintenues constantes, on peut écrire:
Si la variation d’enthalpie ( H) est positive (réaction endothermique), on voit que ln KN et, par suite, KN croissent par élévation de température, ce qui ne peut se faire que si L, M augmentent et si A, B diminuent: ainsi, une élévation de température déplace l’équilibre dans le sens endothermique.Influence de la pression et du volumeLes variables autres que la pression étant maintenues constantes, on peut écrire: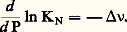 Cette relation est généralement attribuée à Le Chatelier.Un raisonnement analogue au précédent montre que 漣 益 joue dans cette discussion le même rôle que H: un accroissement de la pression, déplace l’équilibre dans le sens de la diminution du nombre de molécules.Ainsi, dans la synthèse de l’ammoniac:
Cette relation est généralement attribuée à Le Chatelier.Un raisonnement analogue au précédent montre que 漣 益 joue dans cette discussion le même rôle que H: un accroissement de la pression, déplace l’équilibre dans le sens de la diminution du nombre de molécules.Ainsi, dans la synthèse de l’ammoniac: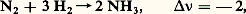 le rendement sera nettement favorisé par l’augmentation de la pression.En ce qui concerne l’influence du volume, étant donné que le volume d’un système gazeux varie en sens inverse de sa pression, l’influence d’un accroissement de volume (à pression constante) sera la même que celle d’une diminution de pression (à volume constant), les autres variables étant maintenues constantes.Ces considérations sont valables pour une solution où l’accroissement de volume est réalisé par addition d’un solvant supposé chimiquement inerte vis-à-vis des espèces dissoutes étudiées. Par exemple, la dissociation d’un électrolyte AB en ses ions, qui donne lieu à un équilibre si l’électrolyte est faible:
le rendement sera nettement favorisé par l’augmentation de la pression.En ce qui concerne l’influence du volume, étant donné que le volume d’un système gazeux varie en sens inverse de sa pression, l’influence d’un accroissement de volume (à pression constante) sera la même que celle d’une diminution de pression (à volume constant), les autres variables étant maintenues constantes.Ces considérations sont valables pour une solution où l’accroissement de volume est réalisé par addition d’un solvant supposé chimiquement inerte vis-à-vis des espèces dissoutes étudiées. Par exemple, la dissociation d’un électrolyte AB en ses ions, qui donne lieu à un équilibre si l’électrolyte est faible: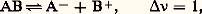 est favorisée par la dilution.Appelons c la concentration globale de l’électrolyte, dissocié ou non, Ka la constante d’action de masse et 見 la fraction dissociée comprise entre 0 et 1; les concentrations de A- et de B+ sont chacune égales à 見c , et celle de AB non dissocié est pratiquement égale à c si l’on admet que 見 est faible. La loi d’action de masse appliquée aux concentrations donne la loi de dilution d’Ostwald:
est favorisée par la dilution.Appelons c la concentration globale de l’électrolyte, dissocié ou non, Ka la constante d’action de masse et 見 la fraction dissociée comprise entre 0 et 1; les concentrations de A- et de B+ sont chacune égales à 見c , et celle de AB non dissocié est pratiquement égale à c si l’on admet que 見 est faible. La loi d’action de masse appliquée aux concentrations donne la loi de dilution d’Ostwald: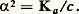 Les constatations précédentes peuvent se résumer dans une proposition qualitative très générale connue sous le nom de principe de Le Chatelier: Toute modification de l’un des facteurs de l’équilibre déplace cet équilibre dans un sens qui tend à s’opposer à la variation du facteur considéré. Ainsi:– Lorsqu’on accroît la température (en fournissant de la chaleur), le système réagit en sens inverse (endothermique);– Lorsqu’on accroît la pression, il tend à s’opposer à cette augmentation, en diminuant le nombre n des molécules (puisque, s’il est gazeux, P = n RT/V);– Lorsqu’on accroît le volume (solution), on diminue la concentration des espèces en solution, et le système réagira dans le sens de leur accroissement...Influence des proportions au départLes mêmes considérations laissent penser qu’en augmentant la proportion d’un des réactifs de départ, B par exemple, le système réagira dans le sens de la consommation de celui-ci (sens 轢), comme le montre la formule utilisée: si B croît, il doit en être de même de L et de M pour que la constance de KN soit respectée.Une complication apparaît néanmoins pour les systèmes gazeux évoluant à pression constante: l’introduction de B produit en effet un accroissement de V (puisque P est maintenue constante). Le déplacement d’équilibre sera dû non seulement à l’excès de B (sens 轢), mais également à l’accroissement de V. Les deux facteurs sont concordants avec 益 礪 0, mais se contrarient si 益 麗 0.On peut montrer que, si 益 諒 漣 b , la proportion de A consommée croît avec le rapport moles B/moles A au départ (exemple 2 + 3 H22 NH3, où B = H2 et b = 3); sinon (exemple 3 H2 + 22 NH3, où B = 2, b = 1), elle passe par un maximum pour une certaine valeur de ce rapport, égal à 漣 b /( 益 + b ), (soit 1 dans le dernier exemple, alors que 1/3 correspond au rapport stœchiométrique) et s’annule aux rapports élevés.5. Prévisions des transformationsTransformations adiabatiquesSi une transformation chimique a lieu, elle obéit à l’inégalité de Clausius représentant l’évolution spontanée du système, soit:
Les constatations précédentes peuvent se résumer dans une proposition qualitative très générale connue sous le nom de principe de Le Chatelier: Toute modification de l’un des facteurs de l’équilibre déplace cet équilibre dans un sens qui tend à s’opposer à la variation du facteur considéré. Ainsi:– Lorsqu’on accroît la température (en fournissant de la chaleur), le système réagit en sens inverse (endothermique);– Lorsqu’on accroît la pression, il tend à s’opposer à cette augmentation, en diminuant le nombre n des molécules (puisque, s’il est gazeux, P = n RT/V);– Lorsqu’on accroît le volume (solution), on diminue la concentration des espèces en solution, et le système réagira dans le sens de leur accroissement...Influence des proportions au départLes mêmes considérations laissent penser qu’en augmentant la proportion d’un des réactifs de départ, B par exemple, le système réagira dans le sens de la consommation de celui-ci (sens 轢), comme le montre la formule utilisée: si B croît, il doit en être de même de L et de M pour que la constance de KN soit respectée.Une complication apparaît néanmoins pour les systèmes gazeux évoluant à pression constante: l’introduction de B produit en effet un accroissement de V (puisque P est maintenue constante). Le déplacement d’équilibre sera dû non seulement à l’excès de B (sens 轢), mais également à l’accroissement de V. Les deux facteurs sont concordants avec 益 礪 0, mais se contrarient si 益 麗 0.On peut montrer que, si 益 諒 漣 b , la proportion de A consommée croît avec le rapport moles B/moles A au départ (exemple 2 + 3 H22 NH3, où B = H2 et b = 3); sinon (exemple 3 H2 + 22 NH3, où B = 2, b = 1), elle passe par un maximum pour une certaine valeur de ce rapport, égal à 漣 b /( 益 + b ), (soit 1 dans le dernier exemple, alors que 1/3 correspond au rapport stœchiométrique) et s’annule aux rapports élevés.5. Prévisions des transformationsTransformations adiabatiquesSi une transformation chimique a lieu, elle obéit à l’inégalité de Clausius représentant l’évolution spontanée du système, soit: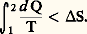 Si, de plus, elle est adiabatique, à tout moment d Q = 0, donc la condition pour que la transformation ait lieu est S 礪 0.On a vu que la condition pour qu’une réaction à température et à pression données ait lieu est que la variation d’énergie libre qui l’accompagne soit négative. Utilisant l’expression de GT,P donnée plus haut et compte tenu de GT,P = 漣 RT ln KP, on déduit la condition:
Si, de plus, elle est adiabatique, à tout moment d Q = 0, donc la condition pour que la transformation ait lieu est S 礪 0.On a vu que la condition pour qu’une réaction à température et à pression données ait lieu est que la variation d’énergie libre qui l’accompagne soit négative. Utilisant l’expression de GT,P donnée plus haut et compte tenu de GT,P = 漣 RT ln KP, on déduit la condition: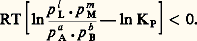 Il faut bien remarquer que les p i désignent maintenant les valeurs actuelles dans le système et non les valeurs à l’équilibre; si G est très négatif, le système est très éloigné de l’équilibre et la transformation sera d’autant plus aisée.Afin de réaliser, par exemple, la réaction:
Il faut bien remarquer que les p i désignent maintenant les valeurs actuelles dans le système et non les valeurs à l’équilibre; si G est très négatif, le système est très éloigné de l’équilibre et la transformation sera d’autant plus aisée.Afin de réaliser, par exemple, la réaction: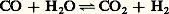 (KP = 0,71 à 700 0C), on introduit dans un réacteur porté à cette température (973 K), renfermant déjà de l’hydrogène et du dioxyde de carbone à des pressions partielles de 1,5 atm., du monoxyde de carbone à 10 atm. et de la vapeur d’eau à 5 atm. Ces deux dernières substances vont-elles réagir?
(KP = 0,71 à 700 0C), on introduit dans un réacteur porté à cette température (973 K), renfermant déjà de l’hydrogène et du dioxyde de carbone à des pressions partielles de 1,5 atm., du monoxyde de carbone à 10 atm. et de la vapeur d’eau à 5 atm. Ces deux dernières substances vont-elles réagir?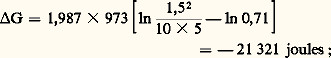 la réaction est donc possible.Si G est positif, c’est évidemment la réaction inverse qui a lieu.Dans une transformation à la température T, il arrive fréquemment que le terme entropique T S de l’expression G = H 漣 T S soit petit par rapport au terme enthalpique (chaleur de réaction), surtout si T est faible; dans ce cas, les conditions G 麗 0 et H 麗 0 sont équivalentes. On retrouve le principe de Berthelot : Si plusieurs réactions sont possibles, la plus exothermique est la plus probable.La condition G 麗 0 est évidemment la seule rigoureuse.Elle est d’ailleurs directement connectée à la notion d’affinité et à celle de variable chimique définies précédemment (formules 16, 17, 18). Lorsqu’on utilise le potentiel chimique molaire (cf. 2, potentiel chimique), ces relations prennent la forme:
la réaction est donc possible.Si G est positif, c’est évidemment la réaction inverse qui a lieu.Dans une transformation à la température T, il arrive fréquemment que le terme entropique T S de l’expression G = H 漣 T S soit petit par rapport au terme enthalpique (chaleur de réaction), surtout si T est faible; dans ce cas, les conditions G 麗 0 et H 麗 0 sont équivalentes. On retrouve le principe de Berthelot : Si plusieurs réactions sont possibles, la plus exothermique est la plus probable.La condition G 麗 0 est évidemment la seule rigoureuse.Elle est d’ailleurs directement connectée à la notion d’affinité et à celle de variable chimique définies précédemment (formules 16, 17, 18). Lorsqu’on utilise le potentiel chimique molaire (cf. 2, potentiel chimique), ces relations prennent la forme: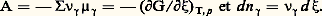 Une intégration entre 﨡 = 0 et 﨡 = 1, ce qui correspond à un équivalent de la réaction, donne la relation cherchée:
Une intégration entre 﨡 = 0 et 﨡 = 1, ce qui correspond à un équivalent de la réaction, donne la relation cherchée: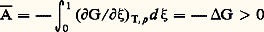 où 漣 G se présente sous la forme d’une valeur moyenne de l’affinité entre le début et la fin de la réaction considérée.Valeur de ces prévisionsSi l’on calcule à température ordinaire la variation d’énergie libre de la réaction:
où 漣 G se présente sous la forme d’une valeur moyenne de l’affinité entre le début et la fin de la réaction considérée.Valeur de ces prévisionsSi l’on calcule à température ordinaire la variation d’énergie libre de la réaction: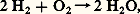 on trouve une valeur fortement négative: la combinaison des deux gaz devrait donc être très facile; or, il n’en est rien, et leur mélange persiste indéfiniment. Inversement, à la température ordinaire, le monoxyde d’azote NO devrait se décomposer en ses éléments:
on trouve une valeur fortement négative: la combinaison des deux gaz devrait donc être très facile; or, il n’en est rien, et leur mélange persiste indéfiniment. Inversement, à la température ordinaire, le monoxyde d’azote NO devrait se décomposer en ses éléments: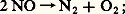 or le monoxyde d’azote obtenu chimiquement par réduction de l’acide nitrique n’évolue pas. En réalité, pour qu’une transformation se produise, la condition G 麗 0 n’est pas la seule; il faut que la vitesse de transformation soit suffisante. S’il n’en est pas ainsi, la réaction n’aura pas lieu, quoiqu’elle soit thermodynamiquement possible; il faut alors rechercher un catalyseur qui, accroissant la vitesse, la réalisera (un catalyseur n’altère pas la configuration finale, il permet de l’atteindre plus rapidement).De toute façon, la connaissance de G est très utile, car un G positif prouve que la réaction est impossible, et cette connaissance évite bien des essais inutiles.On peut constater ainsi que l’étude de la cinétique complète celle de la thermodynamique.Revenons au système H2 + 2 gazeux à la température ordinaire; on peut considérer qu’il est bien en équilibre, puisque le temps ne le modifie pas, cependant il ne constitue pas l’état le plus stable de ce système: on dit qu’il est métastable .La métastabilité peut toujours être rompue par une cause extérieure, en particulier un catalyseur convenable; ainsi, dans le cas de la synthèse de l’eau, une pointe de platine portée au rouge déclenche une réaction violente avec formation d’eau. Ces considérations concernent également les phénomènes physiques: au-dessous de 0 0C, il est possible d’obtenir de l’«eau surfondue», mais cette eau est métastable par rapport à la glace et l’introduction d’un germe de cette dernière provoque la prise en masse.La métastabilité enrichit évidemment la chimie d’un grand nombre d’espèces; sans elle, la chimie organique serait bien pauvre, car la plupart de ses composés sont métastables par rapport au mélange de carbone et d’hydrogène. Ainsi, pour la réaction:
or le monoxyde d’azote obtenu chimiquement par réduction de l’acide nitrique n’évolue pas. En réalité, pour qu’une transformation se produise, la condition G 麗 0 n’est pas la seule; il faut que la vitesse de transformation soit suffisante. S’il n’en est pas ainsi, la réaction n’aura pas lieu, quoiqu’elle soit thermodynamiquement possible; il faut alors rechercher un catalyseur qui, accroissant la vitesse, la réalisera (un catalyseur n’altère pas la configuration finale, il permet de l’atteindre plus rapidement).De toute façon, la connaissance de G est très utile, car un G positif prouve que la réaction est impossible, et cette connaissance évite bien des essais inutiles.On peut constater ainsi que l’étude de la cinétique complète celle de la thermodynamique.Revenons au système H2 + 2 gazeux à la température ordinaire; on peut considérer qu’il est bien en équilibre, puisque le temps ne le modifie pas, cependant il ne constitue pas l’état le plus stable de ce système: on dit qu’il est métastable .La métastabilité peut toujours être rompue par une cause extérieure, en particulier un catalyseur convenable; ainsi, dans le cas de la synthèse de l’eau, une pointe de platine portée au rouge déclenche une réaction violente avec formation d’eau. Ces considérations concernent également les phénomènes physiques: au-dessous de 0 0C, il est possible d’obtenir de l’«eau surfondue», mais cette eau est métastable par rapport à la glace et l’introduction d’un germe de cette dernière provoque la prise en masse.La métastabilité enrichit évidemment la chimie d’un grand nombre d’espèces; sans elle, la chimie organique serait bien pauvre, car la plupart de ses composés sont métastables par rapport au mélange de carbone et d’hydrogène. Ainsi, pour la réaction: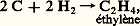 on a G = 68 050 joules.Équilibres non réversiblesIl a été vu que, pour une variation de l’énergie interne négative, le système peut:– réagir complètement (si la réaction est théoriquement complète) ou réagir jusqu’à atteindre la composition relative à l’équilibre (si la réaction est équilibrée);– ne pas réagir du tout si l’évolution prévue est trop lente.Il existe un cas intermédiaire où la réaction a lieu au départ, mais où l’évolution s’arrête avant d’avoir atteint l’état prévu (que la réaction soit complète ou équilibrée). On obtiendra alors, dans le cas d’équilibre, deux limites différentes, selon que l’on part de A + B ou de L + M.Soit, par exemple, la réaction simplifiée A + B 燎 L.Admettons que la proportion de L varie avec la température selon la courbe (C) de la figure 1 si l’équilibre réversible était atteint. On constate, à une température suffisamment basse telle que , l’apparition des deux limites 1 et 2 encadrant la limite Q prévue par la courbe du véritable équilibre qui est représentée en pointillé et qui est déduite des valeurs obtenues aux températures plus élevées par application de l’équation de Van’t Hoff: il n’y a donc plus réversibilité.Les mélanges de compositions comprises entre 1 et 2, ne réagissant pas, sont donc métastables; quant aux courbes lieux de 1 et de 2, ce sont des courbes de faux équilibres .Les vitesses de réaction variant dans le même sens que la température, le freinage précédent, dû à une vitesse insuffisante, doit croître lorsque la température diminue, ce qui se traduit par un élargissement de la zone comprise entre 1 et 2; pour T = , aucune des deux réactions ne devient possible. Ainsi, à la température ordinaire , l’ammoniac est thermodynamiquement stable (100 p. 100 à l’équilibre); de leur côté, l’azote et l’hydrogène devraient réagir pour donner quantitativement de l’ammoniac, mais la figure 1 montre qu’il n’en est rien (cas analogue à celui de H2 + 2).Un composé métastable peut participer à des équilibres réversibles. Ainsi, quoique moins stable que l’ensemble (N2 + 2), le monoxyde d’azote donne lieu, avec le brome, à l’équilibre réversible:
on a G = 68 050 joules.Équilibres non réversiblesIl a été vu que, pour une variation de l’énergie interne négative, le système peut:– réagir complètement (si la réaction est théoriquement complète) ou réagir jusqu’à atteindre la composition relative à l’équilibre (si la réaction est équilibrée);– ne pas réagir du tout si l’évolution prévue est trop lente.Il existe un cas intermédiaire où la réaction a lieu au départ, mais où l’évolution s’arrête avant d’avoir atteint l’état prévu (que la réaction soit complète ou équilibrée). On obtiendra alors, dans le cas d’équilibre, deux limites différentes, selon que l’on part de A + B ou de L + M.Soit, par exemple, la réaction simplifiée A + B 燎 L.Admettons que la proportion de L varie avec la température selon la courbe (C) de la figure 1 si l’équilibre réversible était atteint. On constate, à une température suffisamment basse telle que , l’apparition des deux limites 1 et 2 encadrant la limite Q prévue par la courbe du véritable équilibre qui est représentée en pointillé et qui est déduite des valeurs obtenues aux températures plus élevées par application de l’équation de Van’t Hoff: il n’y a donc plus réversibilité.Les mélanges de compositions comprises entre 1 et 2, ne réagissant pas, sont donc métastables; quant aux courbes lieux de 1 et de 2, ce sont des courbes de faux équilibres .Les vitesses de réaction variant dans le même sens que la température, le freinage précédent, dû à une vitesse insuffisante, doit croître lorsque la température diminue, ce qui se traduit par un élargissement de la zone comprise entre 1 et 2; pour T = , aucune des deux réactions ne devient possible. Ainsi, à la température ordinaire , l’ammoniac est thermodynamiquement stable (100 p. 100 à l’équilibre); de leur côté, l’azote et l’hydrogène devraient réagir pour donner quantitativement de l’ammoniac, mais la figure 1 montre qu’il n’en est rien (cas analogue à celui de H2 + 2).Un composé métastable peut participer à des équilibres réversibles. Ainsi, quoique moins stable que l’ensemble (N2 + 2), le monoxyde d’azote donne lieu, avec le brome, à l’équilibre réversible: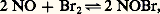 avec production de bromure de nitrosyle.Soit un système porté à une température suffisante pour qu’il se trouve en véritable équilibre. Refroidissons-le très brusquement (trempe) de façon que son équilibre n’ait pas le temps de se déplacer par abaissement de température: on parcourra, sur la figure 1, une horizontale, et l’on aboutira dans la zone de métastabilité, où dorénavant il n’évoluera plus quel que soit le temps. On peut ainsi «figer» le système dans la configuration qu’il avait à température élevée et procéder à son analyse. Il est possible de même d’obtenir à température ordinaire une espèce thermodynamiquement non stable, mais stable à température élevée (cas de l’oxyde FeO au chapitre suivant).6. Exemple d’applicationChoisissons l’exemple des deux équilibres suivants:
avec production de bromure de nitrosyle.Soit un système porté à une température suffisante pour qu’il se trouve en véritable équilibre. Refroidissons-le très brusquement (trempe) de façon que son équilibre n’ait pas le temps de se déplacer par abaissement de température: on parcourra, sur la figure 1, une horizontale, et l’on aboutira dans la zone de métastabilité, où dorénavant il n’évoluera plus quel que soit le temps. On peut ainsi «figer» le système dans la configuration qu’il avait à température élevée et procéder à son analyse. Il est possible de même d’obtenir à température ordinaire une espèce thermodynamiquement non stable, mais stable à température élevée (cas de l’oxyde FeO au chapitre suivant).6. Exemple d’applicationChoisissons l’exemple des deux équilibres suivants: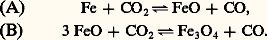 Étude des équilibresPour les deux équilibres, on a:
Étude des équilibresPour les deux équilibres, on a: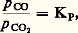 (mais les valeurs de KP sont différentes), ou encore:
(mais les valeurs de KP sont différentes), ou encore: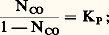 la proportion CO ne dépend que de la température et les courbes I et II représentent ses variations (fig. 2).L’allure de la courbe I montre que l’élévation de la température favorise la réaction dans le sens(NCO croît): la réaction est donc endothermique; c’est l’inverse qui se produit pour la courbe II.Pour ce système, qui implique les mêmes constituants, il est possible d’obtenir deux équilibres où l’on observe successivement l’oxydation, par C2, de Fe en FeO (cas de la courbe I), puis celle de FeO en Fe34 (courbe II), d’où le nom d’équilibres successifs donné à ceux-ci.Les points situés sur les courbes I et II correspondent à des ensembles vérifiant les équilibres (A) ou (B), impliquant deux phases solides (Fe-FeO ou FeO-Fe34). Au contraire, les points situés en dehors de ces courbes ne satisfont plus à ces conditions et sont relatifs à l’existence d’une seule phase. Celle-ci doit être d’autant moins oxydée que le mélange gazeux correspondant à l’un de ces points est plus riche en gaz réducteur (NCO plus élevé); donc, à une température telle que , on doit rencontrer successivement, vers les CO croissants, les domaines de Fe34, de FeO et de Fe.Par exemple, la phase solide rencontrée à la température et pour un mélange gazeux de fraction molaire 0,40 en CO sera du FeO.De plus, on constate que, lorsque la température décroît, le domaine d’existence de FeO s’amenuise pour disparaître vers 565 0C; au-dessous de cette température, on assiste à l’équilibre Fe-Fe34:
la proportion CO ne dépend que de la température et les courbes I et II représentent ses variations (fig. 2).L’allure de la courbe I montre que l’élévation de la température favorise la réaction dans le sens(NCO croît): la réaction est donc endothermique; c’est l’inverse qui se produit pour la courbe II.Pour ce système, qui implique les mêmes constituants, il est possible d’obtenir deux équilibres où l’on observe successivement l’oxydation, par C2, de Fe en FeO (cas de la courbe I), puis celle de FeO en Fe34 (courbe II), d’où le nom d’équilibres successifs donné à ceux-ci.Les points situés sur les courbes I et II correspondent à des ensembles vérifiant les équilibres (A) ou (B), impliquant deux phases solides (Fe-FeO ou FeO-Fe34). Au contraire, les points situés en dehors de ces courbes ne satisfont plus à ces conditions et sont relatifs à l’existence d’une seule phase. Celle-ci doit être d’autant moins oxydée que le mélange gazeux correspondant à l’un de ces points est plus riche en gaz réducteur (NCO plus élevé); donc, à une température telle que , on doit rencontrer successivement, vers les CO croissants, les domaines de Fe34, de FeO et de Fe.Par exemple, la phase solide rencontrée à la température et pour un mélange gazeux de fraction molaire 0,40 en CO sera du FeO.De plus, on constate que, lorsque la température décroît, le domaine d’existence de FeO s’amenuise pour disparaître vers 565 0C; au-dessous de cette température, on assiste à l’équilibre Fe-Fe34: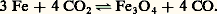 Si FeO peut exister à la température ordinaire, c’est parce qu’il se trouve à l’état métastable (obtenu par trempe); la variation d’énergie libre de la réaction:
Si FeO peut exister à la température ordinaire, c’est parce qu’il se trouve à l’état métastable (obtenu par trempe); la variation d’énergie libre de la réaction: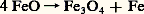 est bien négative, mais la vitesse est quasi nulle.La figure 2 montre qu’à la température Fe34 et FeO seront en équilibre pour une proportion CO correspondant à l’ordonnée du point M. Si CO prend une valeur soit un peu supérieure, soit un peu inférieure à celle-ci, il y aura transformation, respectivement, en FeO ou en Fe34.C’est là un critère de la réversibilité: une modification même faible des conditions opératoires provoque un déplacement sensible de l’équilibre, et des modifications de sens contraire conduisent à des effets inverses.Réduction des oxydes de fer dans un haut fourneauDans un haut fourneau, il y a toujours un excès de carbone (coke), et l’équilibre C2 + C = 2 CO (fig. 2, courbe III, à la pression atmosphérique) doit obligatoirement être réalisé; CO, imposé en principe (cf. supra ), devrait décroître lorsque la température diminue (réaction endothermique).À la partie inférieure, il y a combustion du carbone par injection d’air chaud, et la température est telle (supérieure à 1 000 0C) que CO est très voisin de 1.Les gaz, en s’élevant, traversent des couches de minerai et de coke de moins en moins chaudes; si le système précédent était constamment en équilibre à ces diverses températures, CO devrait diminuer à mesure qu’on s’élève: par exemple, à 600 0C et pour une pression de 1 atm., il serait de 1/3. La figure 2 montre qu’on se trouverait alors dans le domaine d’existence de Fe34, et la réduction en fer métallique ne serait pas possible. Un calcul de la variation d’énergie libre G, fait dans des conditions comparables au calcul donné au chapitre 5, montrerait qu’effectivement cette quantité est positive. En réalité l’équilibre réalisé au bas du fourneau (NCO 黎 1) ne se déplace que très lentement par rapport au temps nécessaire aux gaz pour s’élever à travers les différentes couches. Il s’ensuit que le point figuratif du mélange gazeux se place, en fait, au-dessus de la valeur 1/3 attendue à 600 0C, atteignant une valeur supérieure à celle (0,53) qui correspond à l’équilibre (A). On se trouve alors dans le domaine d’existence du fer, et l’obtention de celui-ci est réellement possible.7. Relations entre grandeurs chimiques et grandeurs électrochimiquesLes piles chimiquesLorsqu’on plonge un métal (du zinc par exemple) dans une solution d’un de ses sels (ions Zn2+), il s’établit une différence de potentiel entre le métal et la solution, qu’on explique par un équilibre métal-ions avec les électrons libres du métal du type:
est bien négative, mais la vitesse est quasi nulle.La figure 2 montre qu’à la température Fe34 et FeO seront en équilibre pour une proportion CO correspondant à l’ordonnée du point M. Si CO prend une valeur soit un peu supérieure, soit un peu inférieure à celle-ci, il y aura transformation, respectivement, en FeO ou en Fe34.C’est là un critère de la réversibilité: une modification même faible des conditions opératoires provoque un déplacement sensible de l’équilibre, et des modifications de sens contraire conduisent à des effets inverses.Réduction des oxydes de fer dans un haut fourneauDans un haut fourneau, il y a toujours un excès de carbone (coke), et l’équilibre C2 + C = 2 CO (fig. 2, courbe III, à la pression atmosphérique) doit obligatoirement être réalisé; CO, imposé en principe (cf. supra ), devrait décroître lorsque la température diminue (réaction endothermique).À la partie inférieure, il y a combustion du carbone par injection d’air chaud, et la température est telle (supérieure à 1 000 0C) que CO est très voisin de 1.Les gaz, en s’élevant, traversent des couches de minerai et de coke de moins en moins chaudes; si le système précédent était constamment en équilibre à ces diverses températures, CO devrait diminuer à mesure qu’on s’élève: par exemple, à 600 0C et pour une pression de 1 atm., il serait de 1/3. La figure 2 montre qu’on se trouverait alors dans le domaine d’existence de Fe34, et la réduction en fer métallique ne serait pas possible. Un calcul de la variation d’énergie libre G, fait dans des conditions comparables au calcul donné au chapitre 5, montrerait qu’effectivement cette quantité est positive. En réalité l’équilibre réalisé au bas du fourneau (NCO 黎 1) ne se déplace que très lentement par rapport au temps nécessaire aux gaz pour s’élever à travers les différentes couches. Il s’ensuit que le point figuratif du mélange gazeux se place, en fait, au-dessus de la valeur 1/3 attendue à 600 0C, atteignant une valeur supérieure à celle (0,53) qui correspond à l’équilibre (A). On se trouve alors dans le domaine d’existence du fer, et l’obtention de celui-ci est réellement possible.7. Relations entre grandeurs chimiques et grandeurs électrochimiquesLes piles chimiquesLorsqu’on plonge un métal (du zinc par exemple) dans une solution d’un de ses sels (ions Zn2+), il s’établit une différence de potentiel entre le métal et la solution, qu’on explique par un équilibre métal-ions avec les électrons libres du métal du type: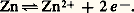 Pour le cuivre, on aurait de même:
Pour le cuivre, on aurait de même: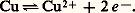 Dans les conditions ordinaires, l’émission d’ions par le métal est très faible, en raison du champ antagoniste intense qui s’établit aussitôt, et s’oppose à leur détachement ultérieur, si bien que le métal reste pratiquement inaltéré. Cependant, si l’on réunit les deux métaux par un fil conducteur et si l’on réalise la jonction des solutions dans lesquelles ils plongent en séparant celles-ci par une paroi poreuse, afin d’éviter leur mélange, les électrons s’écouleront du métal le plus facilement ionisable (le plus électropositif), en l’occurrence le zinc, vers l’autre. On assistera ainsi à un déplacement d’équilibre continuel dans le senspour le premier métal, ou dans le sens 良 pour le second, ce qui se traduira par la réaction:
Dans les conditions ordinaires, l’émission d’ions par le métal est très faible, en raison du champ antagoniste intense qui s’établit aussitôt, et s’oppose à leur détachement ultérieur, si bien que le métal reste pratiquement inaltéré. Cependant, si l’on réunit les deux métaux par un fil conducteur et si l’on réalise la jonction des solutions dans lesquelles ils plongent en séparant celles-ci par une paroi poreuse, afin d’éviter leur mélange, les électrons s’écouleront du métal le plus facilement ionisable (le plus électropositif), en l’occurrence le zinc, vers l’autre. On assistera ainsi à un déplacement d’équilibre continuel dans le senspour le premier métal, ou dans le sens 良 pour le second, ce qui se traduira par la réaction: L’ensemble réalisé constitue une pile (pile Daniell); la différence de potentiel entre les deux métaux (électrodes) s’appelle force électromotrice (f.é.m.) 劉; la réaction qui s’effectue a pour conséquence la production de courant, en raison du déplacement des électrons. Il y a évidemment une relation immédiate entre intensité débitée et vitesse de réaction.Notion de réversibilitéSi l’on applique aux bornes de la pile précédente une force électromotrice égale et opposée à 劉, on n’observe ni courant ni réaction; le système est donc en équilibre. En revanche, si l’on applique une force électromotrice, égale à 劉 梁 﨎, légèrement différente, on observe, selon le signe de 﨎, un courant sensible dans l’un ou l’autre sens, accompagné d’un déplacement de la réaction dans le sens primitif ou dans le sens inverse (dissolution de cuivre et dépôt zinc). Dans le premier sens, le système continue à fonctionner comme pile; dans le second, où la force électromotrice antagoniste est suffisante pour inverser le sens de circulation des électrons, il fonctionne comme cellule d’électrolyse. Un tel comportement est entièrement conforme aux critères de réversibilité donnés pour une réaction chimique (cf. chap. 6).Il existe en revanche des systèmes où 﨎 doit acquérir, dans l’un ou l’autre sens, une valeur notable pour observer un effet mesurable sur la valeur de l’intensité, donc sur le cours de la réaction (surtension): ces systèmes sont irréversibles.Les courbes de polarisation (cf. ÉLECTROCHIMIE ET ÉLECTROLYSE, fig. 3) traduisent bien ces considérations.Grandeurs chimiques et électrochimiquesLa réaction chimique qui a lieu dans la pile Daniell pourrait tout aussi bien s’effectuer par des moyens chimiques: une lame de zinc trempée dans une solution de sel de cuivre se recouvre de cuivre métallique. Cependant, dans ce dernier cas, la transformation est notoirement irréversible, car son sens (dépôt de zinc) ne peut être changé en modifiant quelque peu les conditions opératoires, alors qu’on y parvient par voie électrochimique. Néanmoins, la variation d’énergie libre est la même dans les deux cas, puisqu’il s’agit des mêmes états initial et final; il doit donc exister des relations entre grandeurs chimiques et électrochimiques.Les définitions de l’enthalpie libre (G = H 漣 TS) et de l’enthalpie (H = E + PV) conduisent à G = E + PV 漣 TS, soit, pour une transformation à température et à pression constantes,
L’ensemble réalisé constitue une pile (pile Daniell); la différence de potentiel entre les deux métaux (électrodes) s’appelle force électromotrice (f.é.m.) 劉; la réaction qui s’effectue a pour conséquence la production de courant, en raison du déplacement des électrons. Il y a évidemment une relation immédiate entre intensité débitée et vitesse de réaction.Notion de réversibilitéSi l’on applique aux bornes de la pile précédente une force électromotrice égale et opposée à 劉, on n’observe ni courant ni réaction; le système est donc en équilibre. En revanche, si l’on applique une force électromotrice, égale à 劉 梁 﨎, légèrement différente, on observe, selon le signe de 﨎, un courant sensible dans l’un ou l’autre sens, accompagné d’un déplacement de la réaction dans le sens primitif ou dans le sens inverse (dissolution de cuivre et dépôt zinc). Dans le premier sens, le système continue à fonctionner comme pile; dans le second, où la force électromotrice antagoniste est suffisante pour inverser le sens de circulation des électrons, il fonctionne comme cellule d’électrolyse. Un tel comportement est entièrement conforme aux critères de réversibilité donnés pour une réaction chimique (cf. chap. 6).Il existe en revanche des systèmes où 﨎 doit acquérir, dans l’un ou l’autre sens, une valeur notable pour observer un effet mesurable sur la valeur de l’intensité, donc sur le cours de la réaction (surtension): ces systèmes sont irréversibles.Les courbes de polarisation (cf. ÉLECTROCHIMIE ET ÉLECTROLYSE, fig. 3) traduisent bien ces considérations.Grandeurs chimiques et électrochimiquesLa réaction chimique qui a lieu dans la pile Daniell pourrait tout aussi bien s’effectuer par des moyens chimiques: une lame de zinc trempée dans une solution de sel de cuivre se recouvre de cuivre métallique. Cependant, dans ce dernier cas, la transformation est notoirement irréversible, car son sens (dépôt de zinc) ne peut être changé en modifiant quelque peu les conditions opératoires, alors qu’on y parvient par voie électrochimique. Néanmoins, la variation d’énergie libre est la même dans les deux cas, puisqu’il s’agit des mêmes états initial et final; il doit donc exister des relations entre grandeurs chimiques et électrochimiques.Les définitions de l’enthalpie libre (G = H 漣 TS) et de l’enthalpie (H = E + PV) conduisent à G = E + PV 漣 TS, soit, pour une transformation à température et à pression constantes, Si, de plus, la transformation est réversible, on peut écrire T S = Qr (Th. 7) et, puisque E = Qr 漣 Wr , on déduit:
Si, de plus, la transformation est réversible, on peut écrire T S = Qr (Th. 7) et, puisque E = Qr 漣 Wr , on déduit: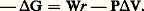 Ainsi, Wr n’incluant pas uniquement le travail P V de la pression, 漣 G est égal au travail réversible des forces autres que la pression, donc ici à celui des forces électriques. Or ce dernier est égal au produit d’une différence de potentiel 劉 par une charge; puisque G est rapporté au nombre de moles de la réaction chimique, la charge doit se rapporter au nombre n 杻 de faradays (1 faraday = 96 500 coulombs) impliqués dans le mécanisme (cf. PILES ET ACCUMULATEURS, chap. 3): ici, on a n = 2 puisque le passage ZnZn2+ fait intervenir deux électrons. On obtient ainsi en valeur absolue:
Ainsi, Wr n’incluant pas uniquement le travail P V de la pression, 漣 G est égal au travail réversible des forces autres que la pression, donc ici à celui des forces électriques. Or ce dernier est égal au produit d’une différence de potentiel 劉 par une charge; puisque G est rapporté au nombre de moles de la réaction chimique, la charge doit se rapporter au nombre n 杻 de faradays (1 faraday = 96 500 coulombs) impliqués dans le mécanisme (cf. PILES ET ACCUMULATEURS, chap. 3): ici, on a n = 2 puisque le passage ZnZn2+ fait intervenir deux électrons. On obtient ainsi en valeur absolue: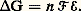 ConséquencesDe d G = 漣 S d T + V d P on déduit:
ConséquencesDe d G = 漣 S d T + V d P on déduit: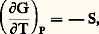 ce qui donne, appliqué aux états initial et final:
ce qui donne, appliqué aux états initial et final: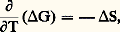 ou:
ou: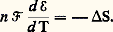 Donc, dans une pile fonctionnant de manière isotherme et réversible, la variation de la force électromotrice avec la température est liée directement à l’entropie de réaction et à la chaleur de réaction effectuée par voie réversible T S. Ainsi, une pile réversible doit, pour fonctionner de façon isotherme, emprunter ou restituer de la chaleur, selon le signe de d 劉/d T, à la source extérieure qui maintient la constance de la température.Si le terme entropique de l’équation G = H 漣 T S est négligeable, on a:
Donc, dans une pile fonctionnant de manière isotherme et réversible, la variation de la force électromotrice avec la température est liée directement à l’entropie de réaction et à la chaleur de réaction effectuée par voie réversible T S. Ainsi, une pile réversible doit, pour fonctionner de façon isotherme, emprunter ou restituer de la chaleur, selon le signe de d 劉/d T, à la source extérieure qui maintient la constance de la température.Si le terme entropique de l’équation G = H 漣 T S est négligeable, on a: formule anciennement utilisée pour relier 劉 à la chaleur de réaction. Elle appelle les mêmes réserves que le principe de Berthelot invoqué plus haut pour prévoir les possibilités de réactions, puisqu’elle néglige les mêmes grandeurs.
formule anciennement utilisée pour relier 劉 à la chaleur de réaction. Elle appelle les mêmes réserves que le principe de Berthelot invoqué plus haut pour prévoir les possibilités de réactions, puisqu’elle néglige les mêmes grandeurs.
Encyclopédie Universelle. 2012.